Publications
Spatially informed cell type deconvolution for spatial transcriptomics
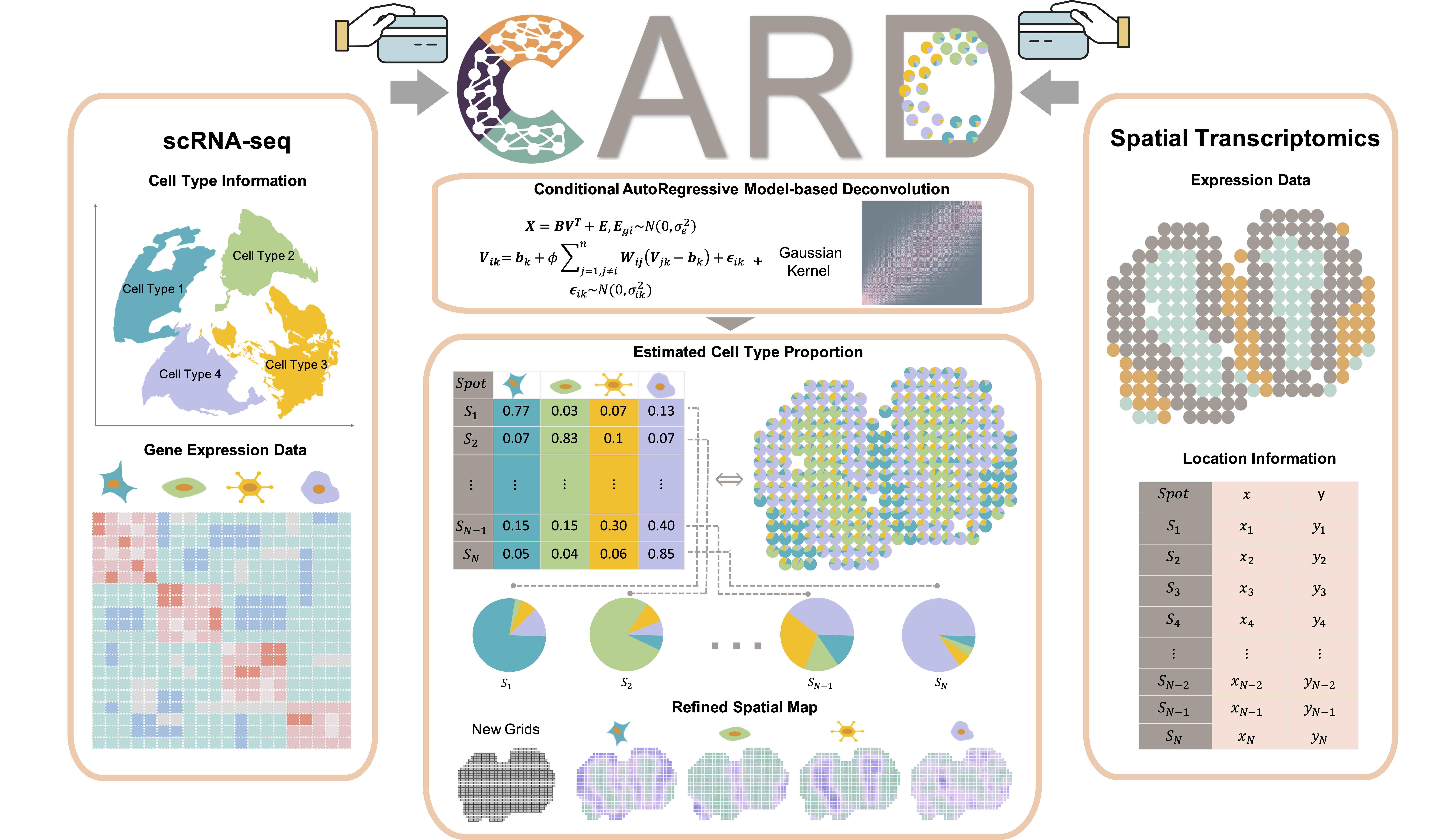
Many spatially resolved transcriptomic technologies do not have single-cell resolution but measure the average gene expression for each spot from a mixture of cells of potentially heterogeneous cell types. Here, we introduce a deconvolution method, conditional autoregressive-based deconvolution (CARD), that combines cell-type-specific expression information from single-cell RNA sequencing (scRNA-seq) with correlation in cell-type composition across tissue locations. Modeling spatial correlation allows us to borrow the cell-type composition information across locations, improving accuracy of deconvolution even with a mismatched scRNA-seq reference. CARD can also impute cell-type compositions and gene expression levels at unmeasured tissue locations to enable the construction of a refined spatial tissue map with a resolution arbitrarily higher than that measured in the original study and can perform deconvolution without an scRNA-seq reference. Applications to four datasets, including a pancreatic cancer dataset, identified multiple cell types and molecular markers with distinct spatial localization that define the progression, heterogeneity and compartmentalization of pancreatic cancer.
Ying Ma and Xiang Zhou
Integrative differential expression and gene set enrichment analysis using summary statistics for scRNA-seq studies
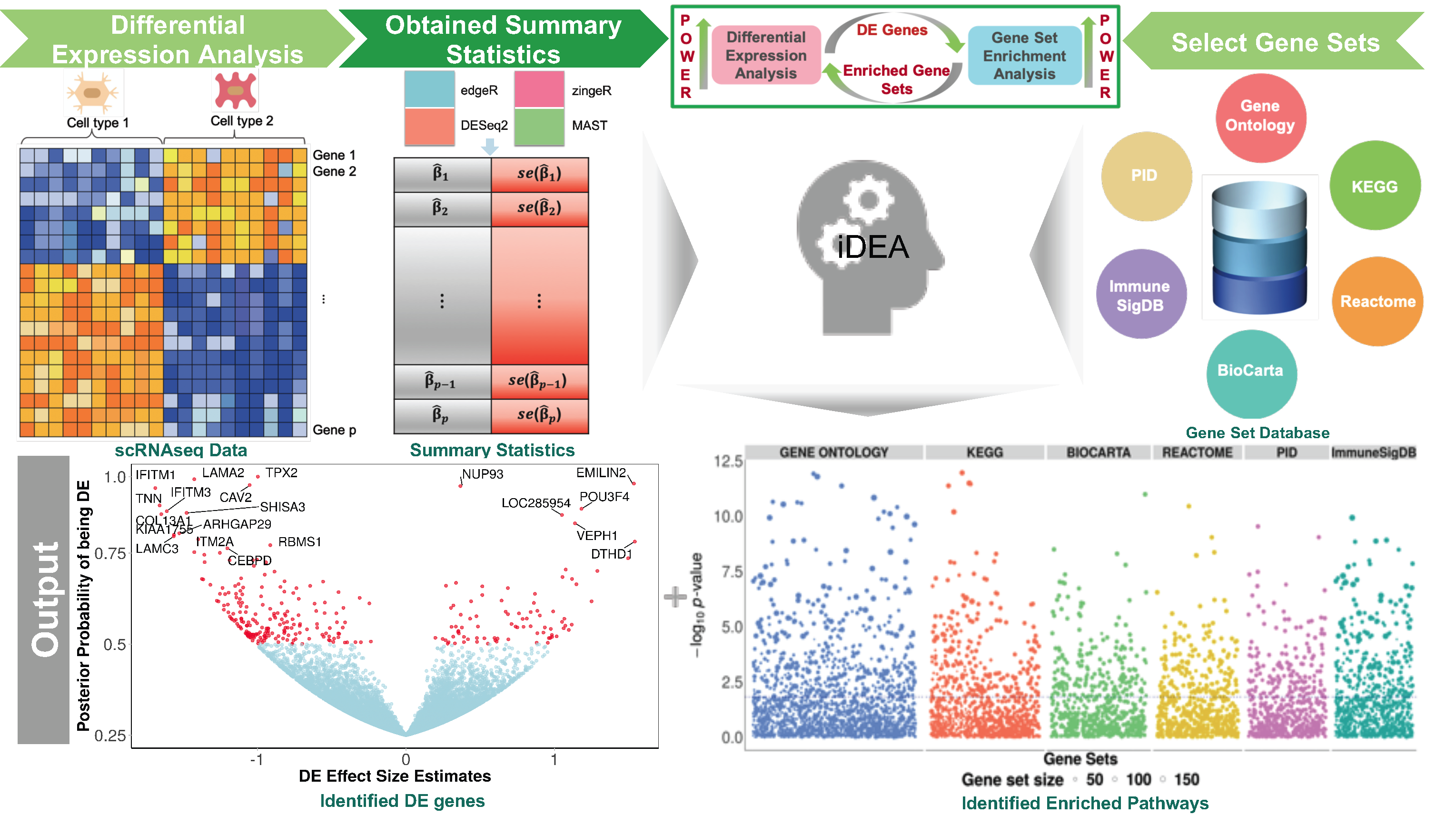
Differential expression (DE) analysis and gene set enrichment (GSE) analysis are commonly applied in single cell RNA sequencing (scRNA-seq) studies. Here, we develop an integrative and scalable computational method, iDEA, to perform joint DE and GSE analysis through a hierarchical Bayesian framework. By integrating DE and GSE analyses, iDEA can improve the power and consistency of DE analysis and the accuracy of GSE analysis. Importantly, iDEA uses only DE summary statistics as input, enabling effective data modeling through complementing and pairing with various existing DE methods. We illustrate the benefits of iDEA with extensive simulations. We also apply iDEA to analyze three scRNA-seq data sets, where iDEA achieves up to five-fold power gain over existing GSE methods and up to 64% power gain over existing DE methods
Ying Ma, Shiquan Sun, Xuequn Shang, Evan T Keller, Mengjie Chen, Xiang Zhou
Accuracy, robustness and scalability of dimensionality reduction methods for single-cell RNA-seq analysis
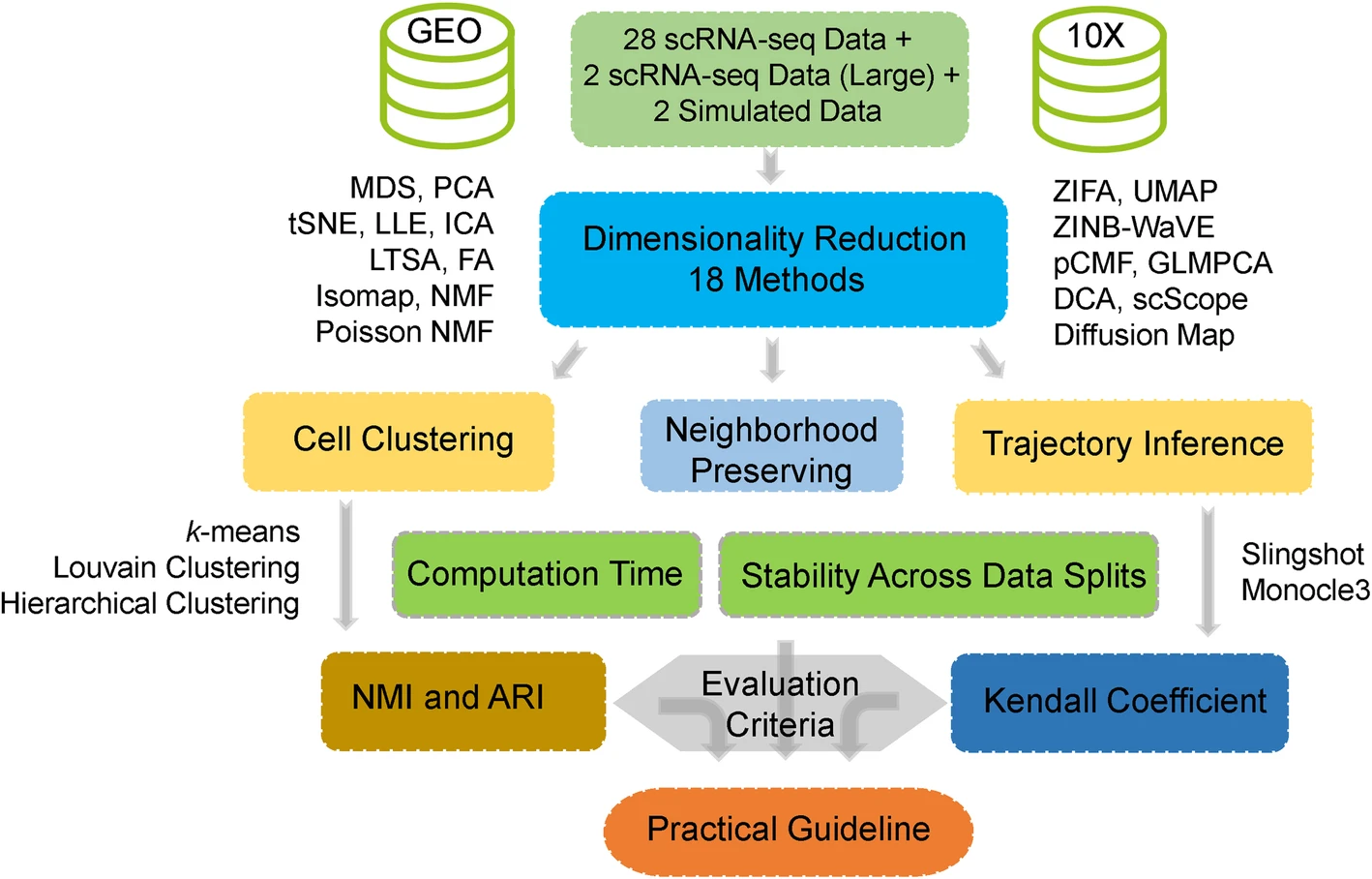
Dimensionality reduction is an indispensable analytic component for many areas of single-cell RNA sequencing (scRNA-seq) data analysis. Proper dimensionality reduction can allow for effective noise removal and facilitate many downstream analyses that include cell clustering and lineage reconstruction. Unfortunately, despite the critical importance of dimensionality reduction in scRNA-seq analysis and the vast number of dimensionality reduction methods developed for scRNA-seq studies, few comprehensive comparison studies have been performed to evaluate the effectiveness of different dimensionality reduction methods in scRNA-seq. We aim to fill this critical knowledge gap by providing a comparative evaluation of a variety of commonly used dimensionality reduction methods for scRNA-seq studies. Specifically, we compare 18 different dimensionality reduction methods on 30 publicly available scRNA-seq datasets that cover a range of sequencing techniques and sample sizes. We evaluate the performance of different dimensionality reduction methods for neighborhood preserving in terms of their ability to recover features of the original expression matrix, and for cell clustering and lineage reconstruction in terms of their accuracy and robustness. We also evaluate the computational scalability of different dimensionality reduction methods by recording their computational cost. Based on the comprehensive evaluation results, we provide important guidelines for choosing dimensionality reduction methods for scRNA-seq data analysis.
Shiquan Sun, Jiaqiang Zhu, Ying Ma, Xiang Zhou
List of publications
Accurate and Efficient Integrative Reference-Informed Spatial Domain Detection for Spatial Transcriptomics
Ying Ma and Xiang Zhou
Nature Methods in press 2024
ExPRSweb: An online repository with polygenic risk scores for common health-related exposures
Ying Ma, Snehal Patil, Xiang Zhou, Bhramar Mukherjee, Lars G Fritsche
The American Journal of Human Genetics 2022
Modeling zero inflation is not necessary for spatial transcriptomics
Peiyao Zhao, Jiaqiang Zhu, Ying Ma, Xiang Zhou
Genome Biology 2022
Spatially informed cell type deconvolution for spatial transcriptomics
Ying Ma and Xiang Zhou
Nature Biotechnology 2022
Genetic prediction of complex traits with polygenic scores: a statistical review
Ying Ma and Xiang Zhou
Trends in Genetics 2021
On cross-ancestry cancer polygenic risk scores
Lars G Fritsche, Ying Ma, Daiwei Zhang, Maxwell Salvatore, Seunggeun Lee, Xiang Zhou, Bhramar Mukherjee
PLoS genetics 2021
Cancer PRSweb: an online repository with polygenic risk scores for major cancer traits and their evaluation in two independent biobanks
Lars G Fritsche, Snehal Patil, Lauren J Beesley, Peter VandeHaar, Maxwell Salvatore, Ying Ma, Robert B Peng, Daniel Taliun, Xiang Zhou, Bhramar Mukherjee
The American Journal of Human Genetics 2021
Integrative differential expression and gene set enrichment analysis using summary statistics for scRNA-seq studies
Ying Ma, Shiquan Sun, Xuequn Shang, Evan T Keller, Mengjie Chen, Xiang Zhou
Nature communications 2020
Accuracy, robustness and scalability of dimensionality reduction methods for single-cell RNA-seq analysis
Shiquan Sun, Jiaqiang Zhu, Ying Ma, Xiang Zhou
Genome Biology 2019