
spMOCA
Background
The rapid advancement of spatially resolved transcriptomics (SRT) technology enables gene expression profiling across tissue locations while preserving spatial context. Gene co-expression analysis in SRT data provides critical insights into how genes function together within the tissue microenvironment. However, existing methods fail to effectively capture the joint influence of gene–gene interactions and spatial dependencies, limiting their biological interpretability.
Overview of spMOCA
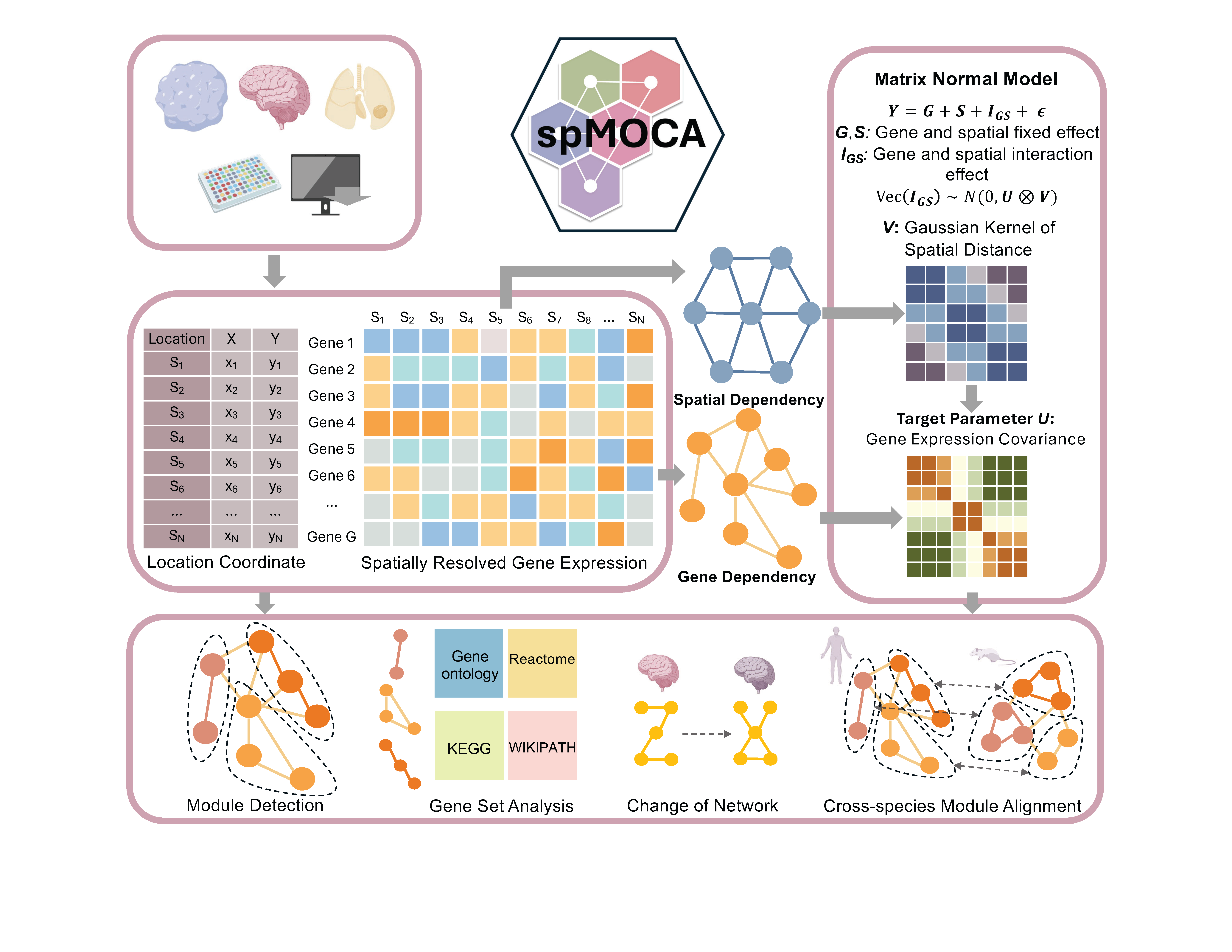
spMOCA is a spatially informed statistical model developed to infer gene co-expression networks across spatial locations in a biologically meaningful way. spMOCA builds upon the principle that gene expression in spatial transcriptomics data is influenced by both spatial dependency and gene-gene interaction. Using a count matrix as input, spMOCA integrates these elements to yield a more accurate and nuanced understanding of gene co-expression networks with spatial contexts. spMOCA employs an efficient optimization algorithm for estimating gene-gene correlation, which is scalable to datasets with tens of thousands of spatial locations and tens of thousands of genes, surpassing the capabilities of existing methods.
Paper
A spatially informed matrix normal model for gene co-expression analysis in spatial transcriptomics studies
Paper Published in Nucleic Acids Research
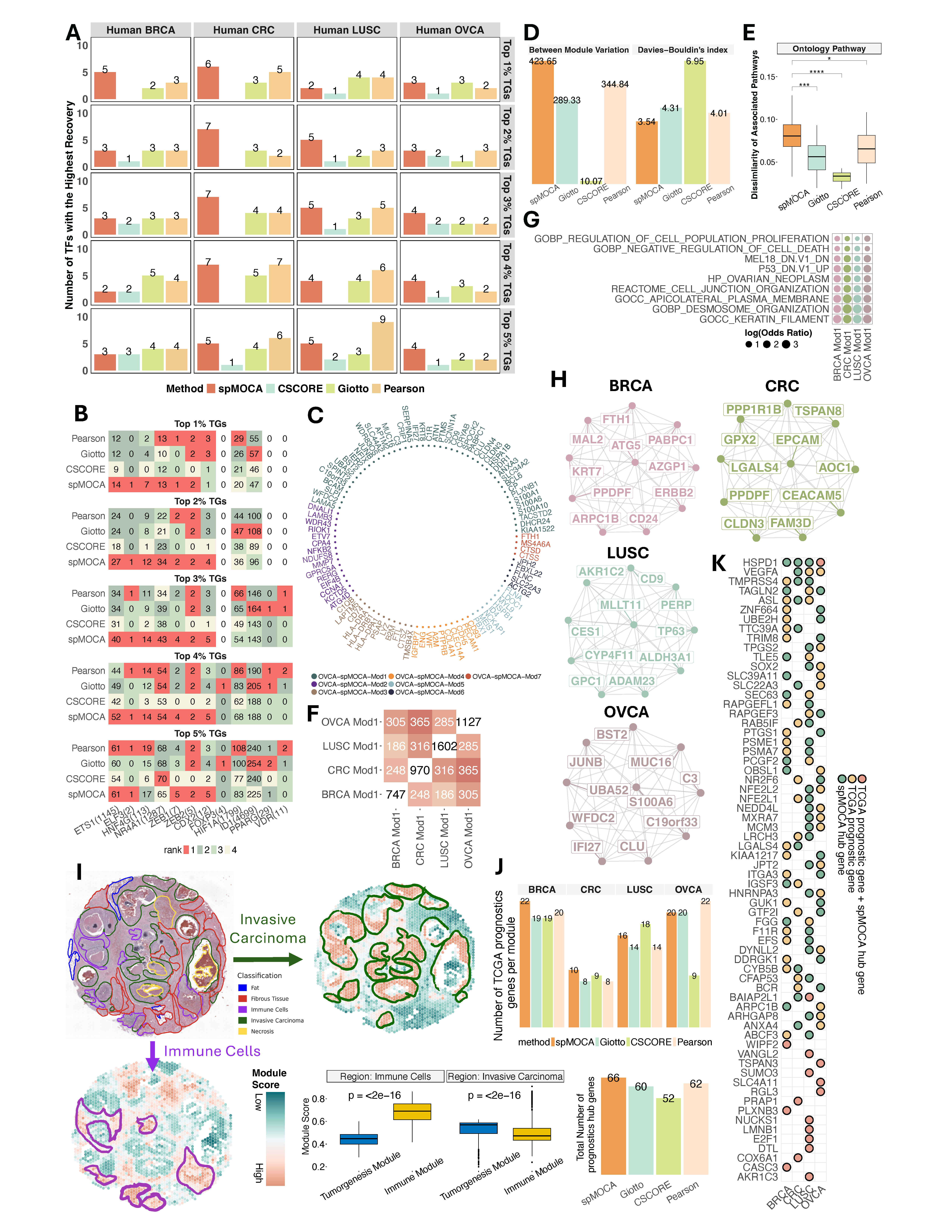
Selected important results by spMOCA
Tutorial of spMOCA
We provided a tutorial for running spMOCA in details, with all kinds of visualization plots provided. Package Website: spMOCA Tutorial